



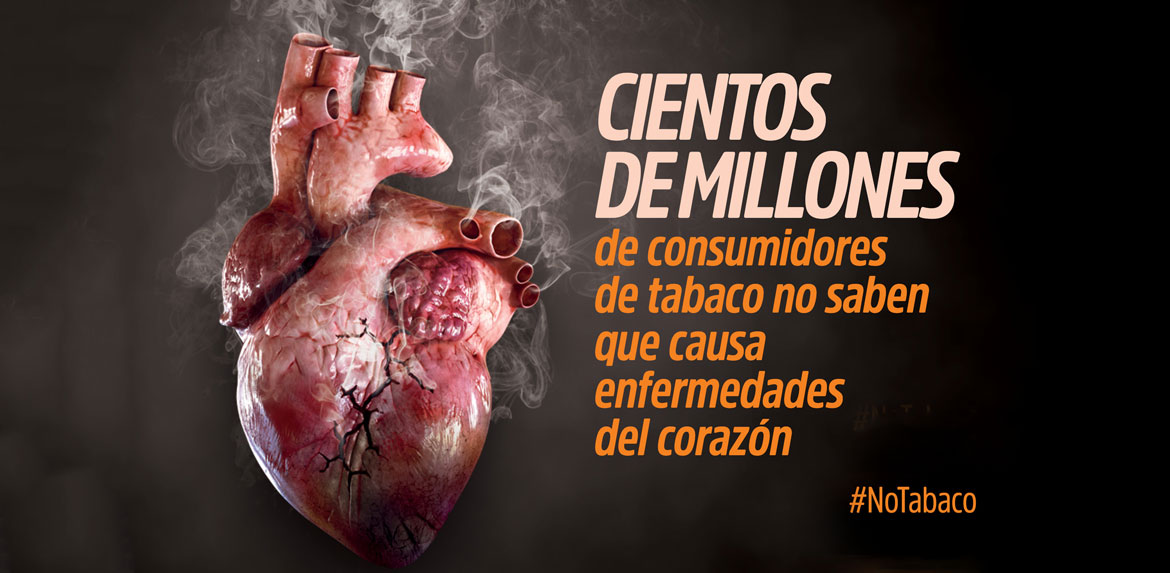







La enfermedad de Gaucher es un trastorno metabólico crónico, progresivo, de origen hereditario, causada por mutaciones en el gen GBA (1q21).
La enfermedad de Gaucher es el trastorno más común de un grupo de más de 40 enfermedades clasificadas como trastornos de almacenamiento lisosómico. Se debe a una deficiencia de una enzima denominada glucocerebrosidasa. Esta enzima degrada y recicla ciertos lípidos llamados glucocerebrósidos. La inactividad de la enzima causa la acumulación de glucocerebrósidos en médula ósea, bazo, hígado, pulmones y cerebro.
En conjunto, los trastornos de almacenamiento lisosómico afectan a 1 de cada 7,700 nacidos vivos. Se estima que aproximadamente 1 persona de cada 40.000 a 60.000 tiene enfermedad de Gaucher y, aunque puede presentarse en grupos de cualquier nacionalidad y origen étnico, es más común en judíos askenazíes (1 de cada 450 personas).
Manifestaciones clínicas
El espectro sintomático de esta enfermedad es amplio y varía desde síntomas muy leves o ausentes a síntomas graves.
- El sistema óseo se afecta con gran frecuencia y es la causa más
frecuente de discapacidad: dolor, fracturas, anomalías esqueléticas
- Aumenta el tamaño de los órganos (hígado, bazo) donde se acumulan
los lípidos
- Se compromete la sangre. Hay anemia con cansancio excesivo, y la
coagulación se altera por disminución de las plaquetas con aparición de hematomas y hemorragias.
- El compromiso neurológico se expresa por retraso mental y demencia,
depresión, síntomas de la enfermedad de Parkinson y afectación de los nervios periféricos (síndrome del túnel carpiano, neuropatía periférica)
- Se observan manchas amarillas y movimientos anormales en los ojos.
- Con frecuencia hay retraso del crecimiento o de la pubertad
- A nivel pulmonar se describen infecciones recurrentes, disnea.
progresiva con insuficiencia respiratoria, y compromiso del intersticio pulmonar. la complicación mas frecuente es la hipertensión pulmonar, más frecuente en mujeres y pacientes con tratamiento irregular o dosis subóptimas. La hipertensión pulmonar grave se observa en 1% de los pacientes, la de grave leve tiene una incidencia de 30% en pacientes no tratados y de un 7% en pacientes en tratamiento con enzima de reemplazo.
Se reconocen tres tipos de enfermedad de Gaucher
- Tipo 1 se manifiesta en la infancia o edad adulta; representa el 95% de los casos y, en con frecuencia es asintomática. No afecta al cerebro
- Tipo 2 o neurológica aguda infantil es poco frecuente pero más grave. Asocia un daño cerebral importante. Se manifiesta desde el nacimiento o los primeros meses, y es rápidamente progresiva causando la muerte alrededor de los 2 años de edad.
- Tipo 3: se presenta en niños y adolescentes. Tiende a evolucionar en forma crónica y avanza lentamente. Afecta al sistema nervioso central.
Los tipos 2 y 3 de enfermedad de Gaucher tienen compromiso neurológico y son exclusivos de la infancia
Diagnóstico
El diagnóstico se hace midiendo la actividad de la enzima glucocerebrosidasa en los glóbulos blancos y en cultivo de células llamadas fibroblastos. Desde el año 2002, científicos argentinos desarrollaron un método que permite identificar pacientes a partir de gotas de sangre colocadas en un papel de filtro, cuando el resultado es anormal se indica la confirmación en los glóbulos blancos
La determinación del nivel de actividad de la enzima es indispensable para el diagnóstico y esencial antes de iniciar el tratamiento
La quitotriosidasa es una enzima cuya actividad está marcadamente incrementada en estos pacientes. Es el indicador más sensible de cambios en la actividad de la enfermedad y contribuye a establecer un pronóstico y al manejo del tratamiento farmacológico.
Otros estudios necesarios son radiografías simples, resonancia magnética, densitometría y gammagrafía ósea para detectar complicaciones óseas
La ecografía cardiaca con doppler, la radiografía de tórax y el electrocardiograma se utilizan para la pesquisar hipertensión arterial pulmonar
Alrededor de la mitad de los pacientes tienen menos de 10 años en el momento del diagnóstico
El estudio genético molecular es muy importante. La enfermedad de Gaucher tiene una herencia autosómica recesiva lo que conlleva un riesgo de recurrencia en los hermanos de un afectado del 25%. Es importante ofrecer este tipo de exámenes a los familiares asintomáticos en riesgo
Tratamiento
Terapia de sustitución enzimática: El Consenso para la Enfermedad de Gaucher del Grupo Argentino de diagnóstico y tratamiento de la Enfermedad de Gaucher del año 2013 establece como el estándar actual el tratamiento de sustitución enzimática. Las sustancias aprobadas a tal fin son imiglucerasa, velaglucerasa-alfa y taliglucerasa-alfa. Este tratamiento mejora significativamente las manifestaciones no neurológicas de la enfermedad tales como anemia, trombopenia, enfermedad ósea, hepatomegalia y esplenomegalia y la calidad de vida de los pacientes con Enfermedad de Gaucher Tipo 1. El tratamiento se aplica por vía intravenosa, es de por vida, individualizado y debe ser dirigido por un equipo multidisciplinario y especializado.
Terapia de reducción de sustratos miglustat y eliglustat inhiben la actividad de la enzima glucocilceramida sintetasa involucrada en la síntesis de los glucolípidos disminuyendo así la producción de glucocerebrósidos y su acumulación en las células. Es una alternativa de segunda línea.
Trasplante de Células Progenitoras Hematopoyéticas ha sido desplazado del tratamiento de la enfermedad de Gaucher tipo 1 por la terapia de sustitución enzimática. Este tratamiento tiene indicación experimental en pacientes con enfermedad de Gaucher tipo 3 sin deterioro neurológico grave severo con progresión de la enfermedad aún con terapia de sustitución enzimática.
Otros tratamientos: control del dolor, transfusiones de sangre, procedimientos ortopédicos. Se recomiendan los bifosfonatos para prevenir las complicaciones óseas derivadas de la osteopenia. La esplenectomía (extirpación del bazo) es una práctica de excepción ya que aumenta la posibilidad de compromiso óseo y pulmonar.

 Calculadoras médicas
Calculadoras médicas  Presentación de casos clínicos
Presentación de casos clínicos  Curso para dejar de fumar
Curso para dejar de fumar